
Cracking the Quantum Scaling Limit with Machine Learned Electron Densities
Authors
Joshua A. Rackers, Lucas Tecot, Mario Geiger, Tess E. Smidt
Abstract
A long-standing goal of science is to accurately solve the Schrödinger equation for large molecular systems. The poor scaling of current quantum chemistry algorithms on classical computers imposes an effective limit of about a few dozen atoms for which we can calculate molecular electronic structure. We present a machine learning (ML) method to break through this scaling limit and make quantum chemistry calculations of very large systems possible. We show that Euclidean Neural Networks can be trained to predict the electron density with high fidelity from limited data. Learning the electron density allows us to train a machine learning model on small systems and make accurate predictions on large ones. We show that this ML electron density model can break through the quantum scaling limit and calculate the electron density of systems of thousands of atoms with quantum accuracy.
Concepts
The Big Picture
Imagine trying to predict the weather over an entire continent, but your supercomputer can only handle a city block at a time. Scale it up and the math explodes, not linearly, but catastrophically. That is precisely the situation chemists and physicists face when simulating large molecules using quantum mechanics.
The equations governing electrons are well-known and exact. The problem is that solving them gets exponentially harder as you add more atoms. The gold-standard method, coupled cluster theory, becomes ferociously expensive as molecules grow: doubling the number of atoms can multiply the required computing time by a factor of 64 or more.
A calculation on a single water molecule takes minutes. A calculation on a modest protein would take longer than the age of the universe.
This “quantum scaling limit” has been one of the most stubborn walls in computational science. It caps rigorous quantum chemistry at around a few dozen atoms, placing entire categories of important problems (protein-drug binding, enzyme design, materials analysis) out of reach. Researchers have developed clever approximations, but approximations sacrifice accuracy. The dream is quantum-level accuracy at biological scales.
A team from Sandia National Laboratories, UCLA, EPFL, and MIT has found a way to break through that wall using machine learning, achieving quantum-accurate electron density predictions for systems of more than 1,000 atoms.
Key Insight: By training a machine learning model to predict the quantum electron density, rather than energies or forces directly, the team exploits a fundamental physical principle that lets them train on small systems and accurately predict behavior in arbitrarily large ones.
How It Works
The breakthrough rests on a 1996 insight from Nobel laureate Walter Kohn: the “nearsightedness of electronic matter.” Electrons don’t care about what’s happening far away. At some finite distance, quantum correlations between electrons decay to zero. The quantum behavior of an electron is shaped almost entirely by its immediate neighborhood, not by atoms thousands of angstroms (each roughly the width of one atom) away.
If that’s true, a model trained on small molecular clusters, capturing the local quantum environment, should generalize to arbitrarily large systems. The team built exactly that model, using water clusters as their test case.
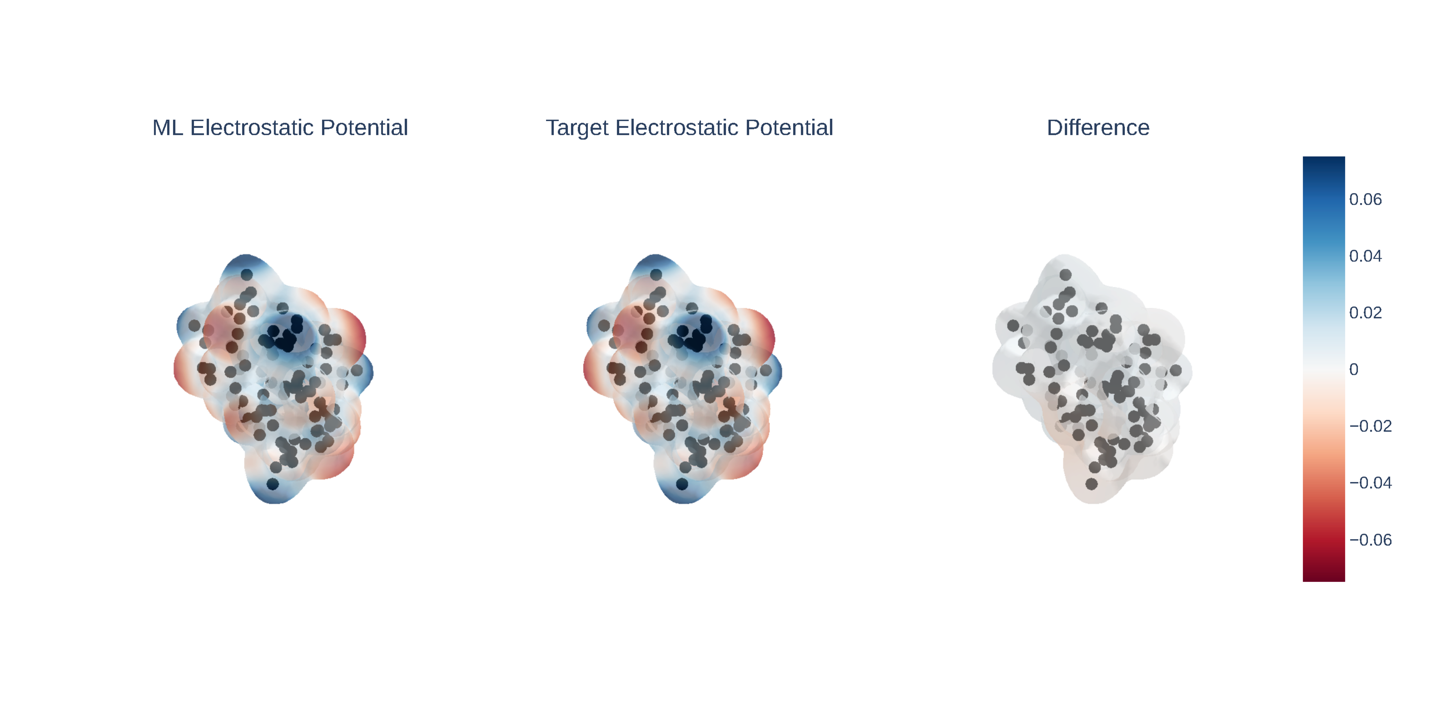
The key technical choice was to predict the electron density itself, not derived properties like energy. The electron density is a three-dimensional map of where electrons are most likely to be found, the most fundamental quantum property of a system, from which everything else can in principle be derived. The researchers represent this map as a sum of bell-shaped mathematical functions centered on each atom, then train the network to predict how strongly each function contributes at each location.
Here is where most machine learning approaches fail. Those basis functions contain spherical harmonics, mathematical functions that encode directional information in 3D space, like lines of longitude and latitude on a globe but applied to electron clouds. Rotate a molecule, and the electron density must rotate with it.
This property is called equivariance: the output transforms consistently with the input. Most neural networks are invariant, meaning outputs don’t change with rotation. That sounds desirable, but for a quantity like electron density that has directionality baked in, invariance is actually a bug.
The team used their own framework, e3nn (Euclidean Neural Networks), built to encode all symmetries of 3D Euclidean space, including rotational equivariance. When tested on rotated versions of a water molecule it had memorized perfectly, an invariant model’s errors exploded with each rotation. The equivariant e3nn model? Zero error, identical output for identical geometry, regardless of orientation.
Equivariance also improves data efficiency. The team trained five networks with increasing spherical harmonic degree, a measure of how much directional detail the model captures. Richer equivariant features required far less data to reach the same accuracy. Since high-level quantum chemistry calculations are expensive, this matters enormously.
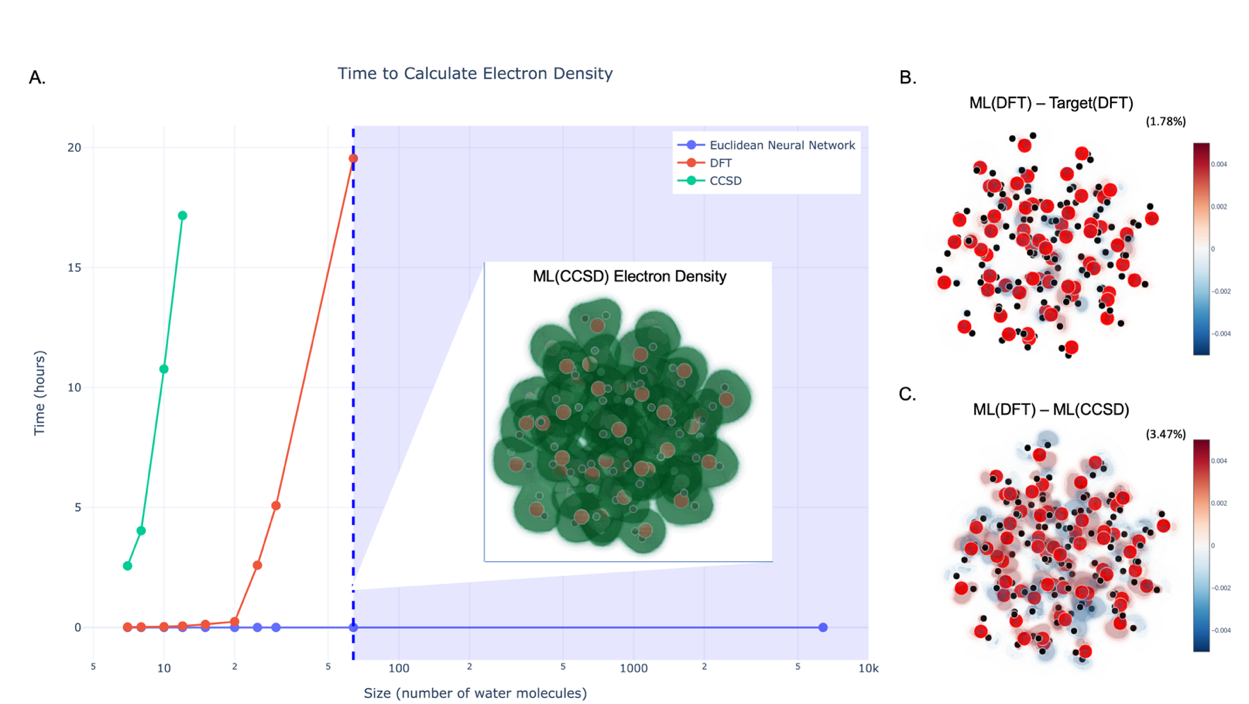
The scaling experiment is where the real payoff shows up:
- The model is trained exclusively on small water clusters
- It is evaluated on progressively larger clusters, up to thousands of atoms
- Prediction accuracy does not degrade as systems grow larger
- Above a certain training cluster size, accuracy plateaus
That plateau is scientifically meaningful on its own. It implies a characteristic length scale beyond which quantum correlations vanish in liquid water, and the machine learning model finds that length scale empirically, without being told what it is.
Why It Matters
Drug discovery, materials science, and biochemistry are all bottlenecked by our inability to run quantum calculations on large molecules. A method that trains on small, tractable systems and accurately predicts large, intractable ones doesn’t just speed things up. It opens entire categories of research that have been structurally impossible.
There is also a deeper AI story here. The equivariant neural network approach behind e3nn is closely related to the architectures used in AlphaFold 2 and RoseTTAFold, the landmark protein structure predictors. Showing that equivariant networks can capture quantum mechanical properties, not just geometric structure, extends this class of models into the heart of physics.
The result points to a broader principle: embed the right physical symmetries into a model and you cut the data needed to learn by orders of magnitude. Nature already knows the rules; good machine learning listens.
Open questions remain. Water is a well-behaved test case; more complex molecules with stronger, longer-range correlations may require larger training sets or more expressive architectures. Extending the approach to predict energies and forces, not just densities, will be the next frontier. But the proof of concept is unambiguous: 1,000+ atoms, quantum accuracy, trained only on small clusters.
Bottom Line: By combining a Nobel-winning physical principle (nearsightedness of electronic matter) with symmetry-aware machine learning (Euclidean Neural Networks), this team has shown that quantum-accurate electron density calculations on thousand-atom systems are not just possible but practical. The full paper is available at arXiv:2201.03726.
IAIFI Research Highlights
This work fuses a foundational concept from quantum physics (nearsightedness of electronic matter) with equivariant machine learning, producing a model that learns quantum mechanics from small systems and applies it at biological scales.
Encoding physical symmetries, specifically rotational equivariance via the e3nn framework, turns out to be essential for learning quantum mechanical quantities, reducing required training data by orders of magnitude compared to invariant approaches.
The model achieves quantum-accurate electron density predictions for systems exceeding 1,000 atoms, breaking through the decades-old quantum scaling limit that has constrained rigorous electronic structure calculations to a few dozen atoms.
Future work will target energy and force predictions, extension to heterogeneous molecular systems, and integration with existing quantum chemistry pipelines; the full paper is available at [arXiv:2201.03726](https://arxiv.org/abs/2201.03726).