
Deep Learning for Bayesian Optimization of Scientific Problems with High-Dimensional Structure
Authors
Samuel Kim, Peter Y. Lu, Charlotte Loh, Jamie Smith, Jasper Snoek, Marin Soljačić
Abstract
Bayesian optimization (BO) is a popular paradigm for global optimization of expensive black-box functions, but there are many domains where the function is not completely a black-box. The data may have some known structure (e.g. symmetries) and/or the data generation process may be a composite process that yields useful intermediate or auxiliary information in addition to the value of the optimization objective. However, surrogate models traditionally employed in BO, such as Gaussian Processes (GPs), scale poorly with dataset size and do not easily accommodate known structure. Instead, we use Bayesian neural networks, a class of scalable and flexible surrogate models with inductive biases, to extend BO to complex, structured problems with high dimensionality. We demonstrate BO on a number of realistic problems in physics and chemistry, including topology optimization of photonic crystal materials using convolutional neural networks, and chemical property optimization of molecules using graph neural networks. On these complex tasks, we show that neural networks often outperform GPs as surrogate models for BO in terms of both sampling efficiency and computational cost.
Concepts
The Big Picture
Imagine designing the perfect light-manipulating material, a photonic crystal engineered at a scale far smaller than a human hair to control which frequencies of light it blocks or transmits. You can’t test every possible arrangement; each simulation takes hours. You need to be smart about which designs to evaluate next. That’s Bayesian optimization: using what you’ve already learned to make educated guesses about where the best answer lies, minimizing expensive experiments.
Scientists have long relied on Gaussian Processes (GPs) to guide this kind of search. A GP is a principled bookkeeper: it tracks not just its best guess about any untested design, but also how confident it is in that guess. The built-in uncertainty estimate is what makes the search smart. It helps the algorithm decide whether to dig deeper into a promising area or venture somewhere entirely new.
GPs have a serious problem, though: they slow to a crawl as inputs grow complex. Design a molecule? GPs struggle. Optimize an image-like grid of material? GPs choke. The more variables you juggle, and the more structure your data has, the worse they perform in both accuracy and speed.
A team from MIT and Google Research has shown a better path: replace the GP with a Bayesian neural network that understands the structure of your data. The result is an optimization engine that handles molecules, images, and high-dimensional physical problems with far greater efficiency.
Key Insight: Bayesian neural networks can encode physical symmetries and structural knowledge as surrogate models, letting researchers run smarter scientific searches in regimes where traditional Gaussian Processes can’t compete.
How It Works
At the heart of any Bayesian optimization loop is the surrogate model: a learned function that predicts how good an untested design will be, along with an uncertainty estimate. That uncertainty is what tells the optimizer whether to exploit a promising region or explore somewhere new.
Traditional GPs compute uncertainty analytically, but at steep cost. Computation scales with the cube of the number of data points. A few hundred experiments? Manageable. A few thousand? Painful. And when inputs are images or molecular graphs rather than simple numerical vectors, you need to hand-engineer a custom kernel function for every new domain.
The team instead turns to Bayesian neural networks (BNNs), which approximate uncertainty through Monte Carlo dropout: randomly disabling neurons during prediction to produce a spread of possible answers that collectively stand in for uncertainty. The key advantage is that any neural network architecture can serve as the surrogate. Convolutional neural networks for image-like inputs, graph neural networks for molecules. The architecture naturally encodes the structure of the problem.
The framework tackles two especially important scenarios:
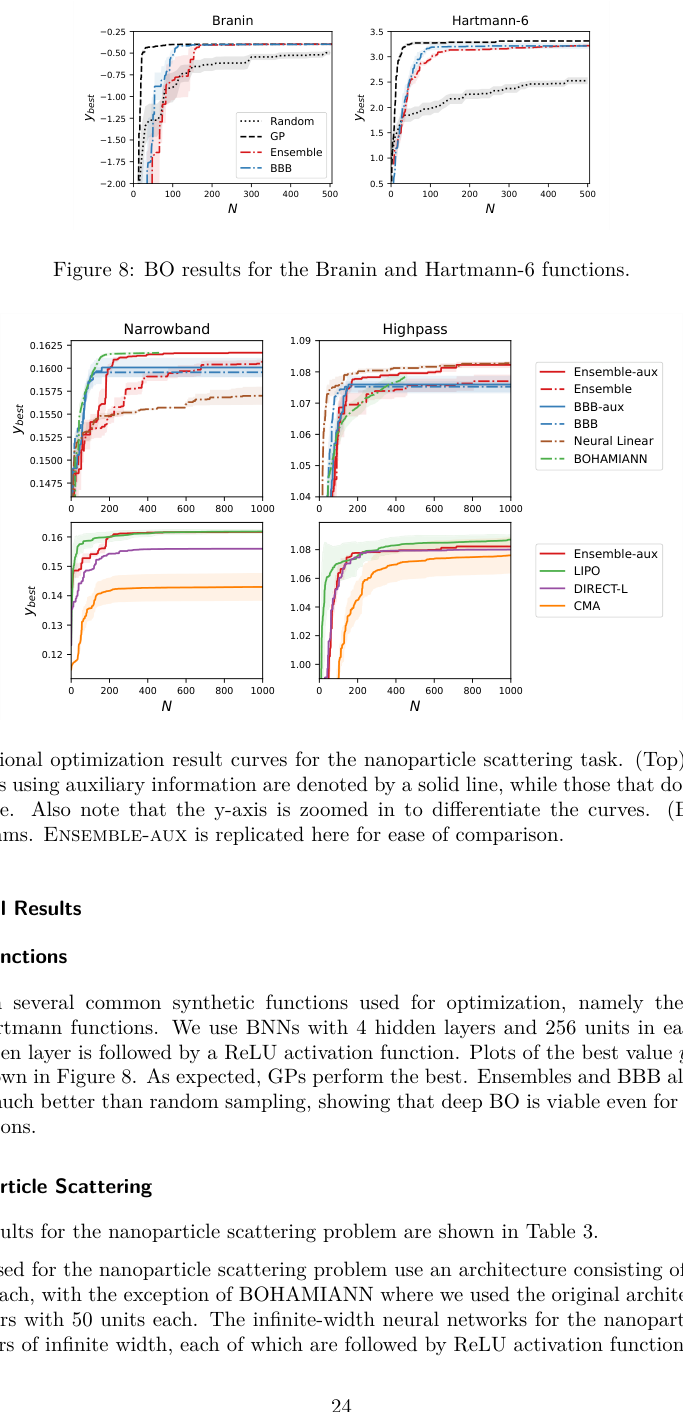
- High-dimensional observations: When an experiment produces rich output (like the full optical scattering spectrum of a nanoparticle across hundreds of wavelengths) the BNN predicts the entire spectrum, then computes the optimization target from it. This auxiliary information sharply improves accuracy compared to predicting a single number.
- Structured input spaces: For photonic crystal design, the input is a binary image, so a convolutional neural network processes it naturally. For molecular design, each molecule is a graph of atoms and bonds, which is exactly what graph neural networks are built to handle.
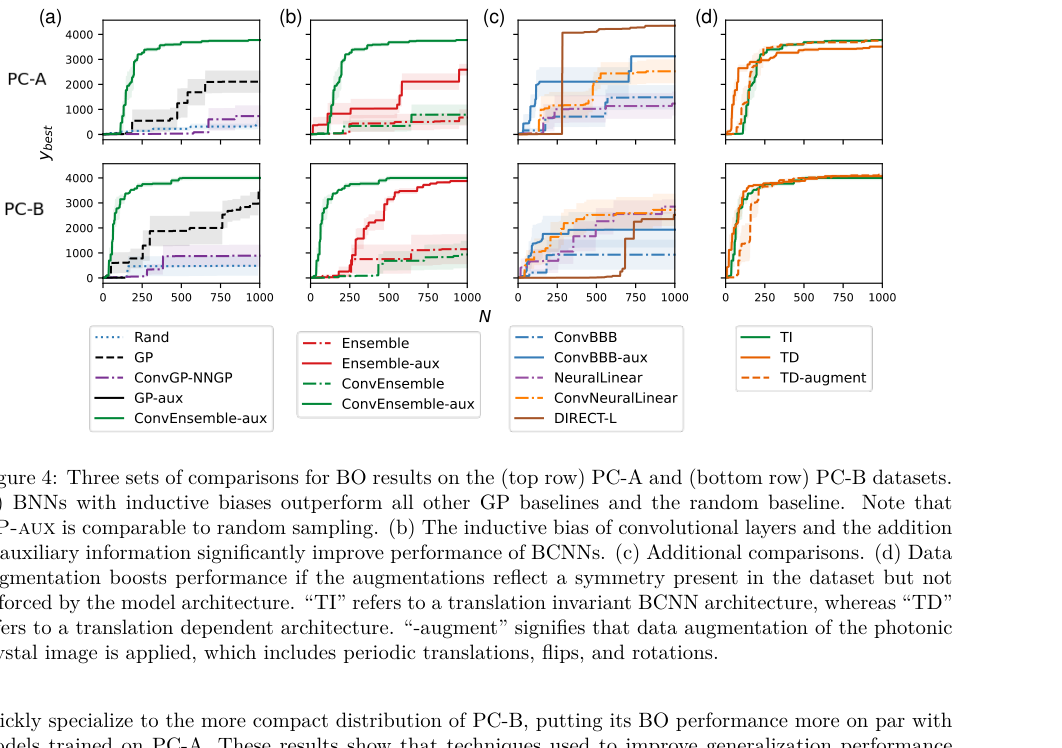
On the photonic crystal task, the goal was to maximize the photonic bandgap, the range of light frequencies the material blocks. The GP surrogate, faced with an image-like input, performed poorly. The CNN-based BNN found much better crystal designs in the same number of evaluations.
For molecule optimization, the team worked with the QM9 dataset, a widely used reference library of small organic molecules with precisely calculated physical and chemical properties. A graph neural network surrogate let the BNN-based optimizer find higher-quality molecules faster than the GP baseline, at a fraction of the computational cost.
Why It Matters
Scientific optimization problems share a common structure, whether they come from drug discovery, materials design, optical engineering, or nuclear physics. Each evaluation is costly, the input space is complex, and there’s structure the optimizer could exploit if its surrogate understood it. GPs were the best available tool for decades, but they were designed for a simpler world.
The deep learning revolution isn’t just about making predictions; it’s about making better decisions. A BNN that understands molecular graphs doesn’t just predict properties. It powers an entire optimization loop in domains that were previously out of reach. As neural architectures keep improving (equivariant networks, transformers, physics-informed models), the surrogates plugged into this framework will only get stronger. The approach also extends to settings with multiple competing objectives or hard physical constraints.
Bottom Line: Replacing Gaussian Processes with domain-aware Bayesian neural networks opens up Bayesian optimization for the complex, high-dimensional, structured problems that arise in real science, while running faster and finding better solutions.
IAIFI Research Highlights
The work brings modern deep learning architectures to bear on practical scientific discovery, applying graph and convolutional neural networks to real optimization tasks in photonics and quantum chemistry.
The research extends Bayesian optimization to high-dimensional structured domains by using BNNs as flexible, scalable surrogates that encode inductive biases inaccessible to traditional Gaussian Processes.
Efficient topology optimization of photonic crystals and quantum chemistry property optimization let researchers explore materials and molecules with specific, physically meaningful properties much faster.
Future extensions could integrate equivariant architectures or multi-objective acquisition functions across computational science; the paper is available at [arXiv:2104.11667](https://arxiv.org/abs/2104.11667).