
High-performance training and inference for deep equivariant interatomic potentials
Authors
Chuin Wei Tan, Marc L. Descoteaux, Mit Kotak, Gabriel de Miranda Nascimento, Seán R. Kavanagh, Laura Zichi, Menghang Wang, Aadit Saluja, Yizhong R. Hu, Tess Smidt, Anders Johansson, William C. Witt, Boris Kozinsky, Albert Musaelian
Abstract
Machine learning interatomic potentials, particularly those based on deep equivariant neural networks, have demonstrated state-of-the-art accuracy and computational efficiency in atomistic modeling tasks like molecular dynamics and high-throughput screening. The size of datasets and demands of downstream workflows are growing rapidly, making robust and scalable software essential. This work presents a major overhaul of the NequIP framework focusing on multi-node parallelism, computational performance, and extensibility. The redesigned framework supports distributed training on large datasets and removes barriers preventing full utilization of the PyTorch 2.0 compiler at train time. We demonstrate this acceleration in a case study by training Allegro models on the SPICE 2 dataset of organic molecular systems. For inference, we introduce the first end-to-end infrastructure that uses the PyTorch Ahead-of-Time Inductor compiler for machine learning interatomic potentials. Additionally, we implement a custom kernel for the Allegro model's most expensive operation, the tensor product. Together, these advancements speed up molecular dynamics calculations on system sizes of practical relevance by up to a factor of 18.
Concepts
The Big Picture
Imagine trying to simulate a drug molecule bouncing around in solution — every bond flexing, every atom jostling its neighbors millions of times per second. To predict whether that molecule will fold correctly, react, or fall apart, you need the forces between every pair of atoms at every instant. Traditional physics-based methods can do this accurately, but they’re agonizingly slow. For years, computational chemists faced an impossible tradeoff: accuracy or speed, but rarely both.
Machine learning changed the rules. AI systems trained on quantum mechanical calculations can now predict atomic forces thousands of times faster than the quantum methods they learned from — without sacrificing accuracy. These systems are called machine learning interatomic potentials (MLIPs). Among the most accurate are equivariant neural networks — models that bake physics directly into their architecture, guaranteeing identical predictions regardless of how you rotate or mirror the atomic system. The NequIP and Allegro models, developed at Harvard and MIT, represent the state of the art.
But as datasets balloon and simulation workflows grow more demanding, even the best models become bottlenecks. A team from Harvard, MIT, and Sandia National Laboratories has now overhauled the NequIP framework from the ground up — and the result is molecular dynamics simulations running up to 18× faster.
Key Insight: By combining modern compiler technology with distributed training and a custom computational kernel, the revamped NequIP framework makes large-scale atomistic simulations practical without sacrificing the physics-grounded accuracy that makes equivariant models so powerful.
How It Works
Three interlocking innovations each target a different bottleneck:
- Compilation — rewriting model code into lean, hardware-optimized instructions
- Distributed training — spreading work intelligently across multiple GPUs
- A custom tensor product kernel — squeezing performance from the model’s most expensive operation
Compilation. PyTorch 2.0 introduced TorchInductor, a compiler that rewrites neural network code into hardware-specific instructions by fusing operations, reordering loops, and eliminating redundant memory transfers. The problem: equivariant models compute atomic forces via automatic differentiation — where the model calculates its own mathematical derivatives on the fly — and that process isn’t a simple primitive operation compilers know how to handle.
The team’s solution was to trace the entire model — the forward prediction pass and the derivative computation together — into a single computational graph using PyTorch’s torch.fx library. Because this combined graph contains only primitive operations, TorchInductor can compile it as a unified whole, enabling aggressive kernel fusion (combining multiple GPU operations into one to avoid costly memory transfers) across energy and force computations simultaneously.
Distributed training. Modern MLIP datasets like SPICE 2 are enormous, demanding work spread across multiple GPUs. The team added Distributed Data Parallel (DDP) training via PyTorch Lightning — but with a critical twist. Standard DDP implementations multitask: while the model computes its backward pass (calculating how wrong its predictions were and how much to adjust each parameter), it simultaneously ships those adjustments (gradients) to other GPUs. That multitasking forces TorchInductor to compile the backward pass in fragments, blocking deeper optimization.
The team’s custom DDP approach instead waits until the entire backward pass completes before sending gradients. The compiler now sees training as one uninterrupted sequence of operations, enabling deeper fusion and lower overhead.
Custom tensor product kernel. Allegro’s most expensive operation is the tensor product — pooling information from neighboring atoms while preserving their symmetry properties. Standard PyTorch has no optimized implementation for this. The team wrote a purpose-built GPU kernel for this operation alone, achieving performance no general-purpose compiler could reach.
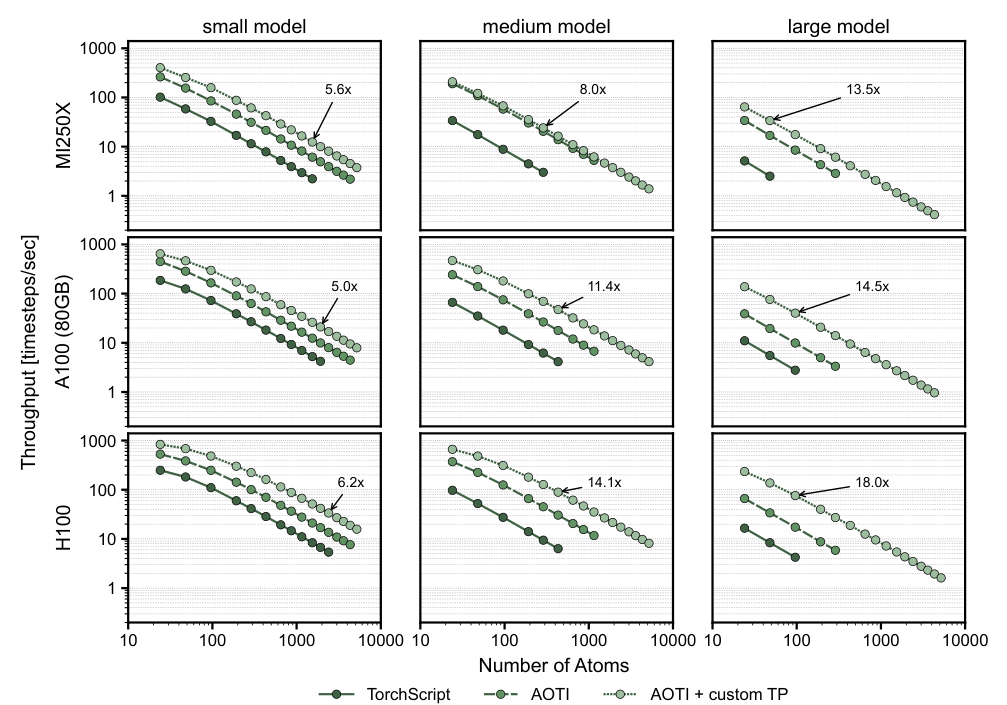
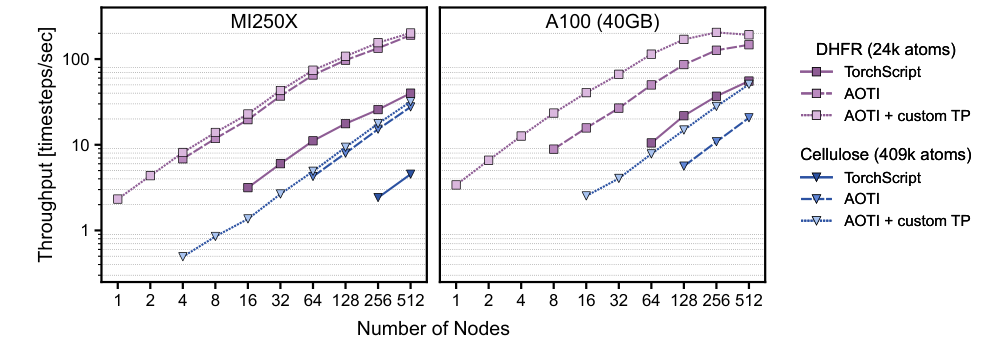
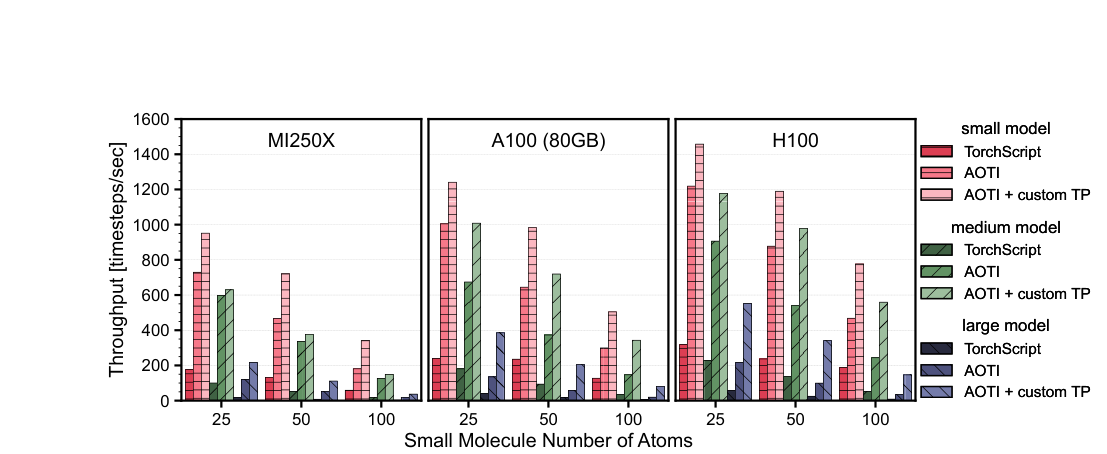
For inference — running the trained model on new atomic configurations — the team introduces the first end-to-end use of Ahead-of-Time Inductor (AOTI) compilation for MLIPs. AOTI compiles models into standalone packages that load directly into C++ simulation codes like LAMMPS, eliminating Python interpreter overhead and replacing the previous TorchScript approach. The compiled model handles dynamic input sizes gracefully, which matters for molecular dynamics, where the number of interacting atom pairs changes every timestep.
Benchmarking on practically relevant system sizes, the new framework achieves up to an 18× speedup in molecular dynamics calculations compared to the previous NequIP release.
Why It Matters
Researchers are racing to build “foundation models” for chemistry — general-purpose potentials trained on millions of diverse molecular configurations and fine-tuned for specific applications. Datasets like SPICE 2, MPTrj, OMat24, and Alexandria collectively span millions of structures. An 18× inference speedup doesn’t just accelerate existing workflows; it opens entirely new ones, from high-throughput drug candidate screening to discovering better battery materials by simulating thousands of candidate chemistries.
The compiler-first approach also closes a longstanding gap. Models developed in Python have historically required heroic engineering to run efficiently in the C++ simulation codes that power large-scale science. By making AOTI compilation a first-class citizen, the NequIP team has shown how Python-developed models can slot cleanly into C++ simulation environments. Other MLIP frameworks (MACE, SevenNet, BAMBOO) currently rely on TorchScript export; the AOTI approach may well become the new standard.
Bottom Line: An 18× speedup in molecular dynamics isn’t incremental progress. It’s the difference between hours and days of compute time, between feasible and infeasible experiments. By taming the compiler and scaling training to modern datasets, this work makes equivariant interatomic potentials ready for the next generation of materials and molecular science.
IAIFI Research Highlights
This work connects advances in deep learning compiler technology to a core challenge in computational physics — simulating atomic interactions fast enough and accurately enough to be scientifically useful at scale.
The team solved a fundamental barrier to applying TorchInductor to physics-informed neural networks by tracing automatic differentiation into a compiler-friendly computational graph, a technique applicable well beyond MLIPs.
Faster, more scalable equivariant potentials accelerate atomistic simulation across materials science and chemistry, enabling studies of ionic conductivity, phonon properties, and defect behavior that require long simulation timescales.
Future work may extend gradient bucketing strategies to larger architectures and bring AOTI compilation to other MLIP frameworks. The paper is available at [arXiv:2504.16068](https://arxiv.org/abs/2504.16068).