
Learning simple heuristic rules for classifying materials based on chemical composition
Authors
Andrew Ma, Marin Soljačić
Abstract
In the past decade, there has been a significant interest in the use of machine learning approaches in materials science research. Conventional deep learning approaches that rely on complex, nonlinear models have become increasingly important in computational materials science due to their high predictive accuracy. In contrast to these approaches, we have shown in a recent work that a remarkably simple learned heuristic rule -- based on the concept of topogivity -- can classify whether a material is topological using only its chemical composition. In this paper, we go beyond the topology classification scenario by also studying the use of machine learning to develop simple heuristic rules for classifying whether a material is a metal based on chemical composition. Moreover, we present a framework for incorporating chemistry-informed inductive bias based on the structure of the periodic table. For both the topology classification and the metallicity classification tasks, we empirically characterize the performance of simple heuristic rules fit with and without chemistry-informed inductive bias across a wide range of training set sizes. We find evidence that incorporating chemistry-informed inductive bias can reduce the amount of training data required to reach a given level of test accuracy.
Concepts
The Big Picture
Imagine you’re a chemist in the 1800s, long before quantum mechanics, trying to figure out which metals conduct electricity. You notice a pattern: sodium, potassium, copper. They share something. You’re building a rule of thumb based on intuition about the periodic table. Now fast-forward to today, where AI systems predict material properties with stunning accuracy. The catch: the neural networks doing the predicting are so complex that no human can explain why they work. Researchers at MIT are asking a different question: can machines discover those simple, intuitive rules the way a chemist might?
The answer is yes. And the rules that emerge are both surprisingly accurate and genuinely illuminating about the underlying chemistry.
Andrew Ma and Marin Soljačić at MIT have shown that machine learning can produce interpretable, one-number-per-element rules that classify materials (from chemical composition alone) as topological (possessing unusual quantum electronic properties that make them candidates for next-generation technologies) or metallic. Their framework embeds knowledge of the periodic table directly into the learning process, making these simple rules more effective with less training data.
Key Insight: You don’t need a black-box neural network to classify materials. A single number assigned to each element, combined through a simple weighted average, can rival far more complex models, especially when the periodic table’s structure guides the learning.
How It Works
The foundation is a concept the authors previously introduced: topogivity, a single number assigned to each element. To classify a material, look up the topogivity of each element present, take a composition-weighted average (weighted by how much of each element appears in the compound), and check the sign. Positive means topological, negative means conventional. No matrix multiplications, no hidden layers, no hyperparameter tuning.
This paper extends the approach to a second task: metallicity classification, predicting whether a compound is a metal from its chemical formula alone. The team defines two types of models:
- The full model: One free parameter per element. A material’s score is a weighted average of its elements’ scores; classification follows from the sign of that average.
- The restricted model: Same idea, but parameters are tied across elements that occupy similar positions in the periodic table, enforcing the chemical intuition that neighboring elements behave similarly.
The restricted model is where the real contribution lives. Rather than treating carbon and silicon as entirely independent, the model shares information between them: they sit in the same group of the periodic table. This is chemistry-informed inductive bias, baking structural knowledge directly into the model architecture. A student who already knows the periodic table needs fewer examples to learn a new pattern than one starting from scratch. The same logic applies here.
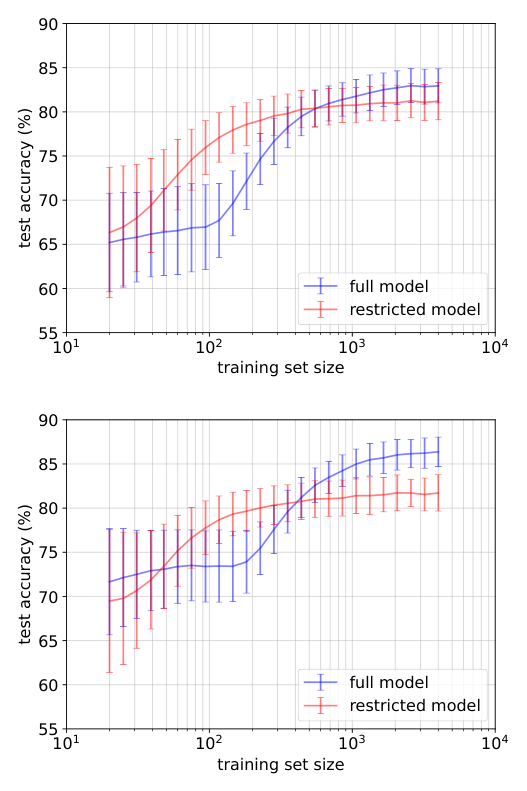
Instead of fitting hundreds of completely independent numbers, the algorithm fits fewer, structured ones. Training uses logistic regression (a standard technique that predicts categories from numerical inputs) with standard regularization, keeping everything interpretable and reproducible. The researchers evaluated both models across training set sizes ranging from just dozens of samples to thousands, on both the topology and metallicity tasks.
Why It Matters
The results are clear: the restricted model consistently reaches a given accuracy level with less training data than the unconstrained full model. In materials science, labeled data is expensive, often requiring ab initio calculations (quantum-mechanics-based simulations built from first principles, without experimental input) or difficult experiments. A model that learns effectively from fewer examples has direct practical value for accelerating materials discovery.
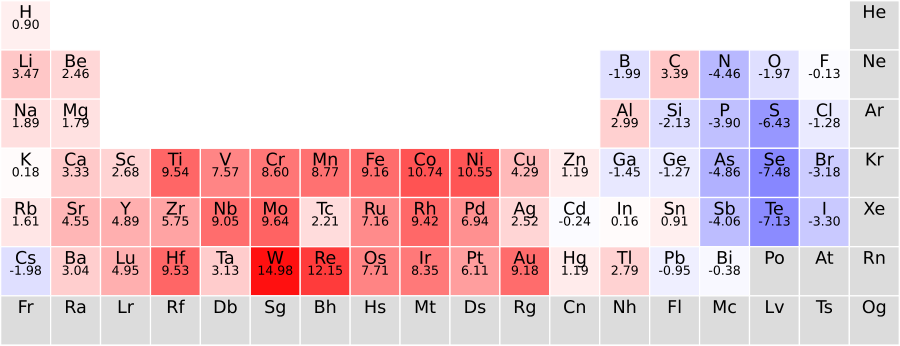
There’s something deeper here about interpretability. The numbers the full model learns for metallicity classification reflect genuine chemical intuition: cesium and francium get high “metallic” scores; oxygen and fluorine score low. The model is rediscovering textbook chemistry from data. That kind of result is rare in modern machine learning, where high accuracy usually comes at the cost of opacity.
This work is part of a broader movement in AI for physical sciences: building models that don’t just predict accurately, but explain. The topogivity approach has already inspired follow-on work, including integration into generative pipelines for discovering new topological materials, compounds that might evade detection by standard symmetry-based methods entirely.
The open questions are worth sitting with. Can this framework extend to properties that depend on crystal structure, not just composition? Can the chemistry-informed inductive bias incorporate electronegativity, atomic radius, or oxidation state trends? As materials databases grow, does the advantage of the restricted model persist, or does built-in physical intuition matter less at scale?
Bottom Line: By learning simple, one-number-per-element rules augmented with periodic table structure, Ma and Soljačić show that interpretable ML can match complex models for materials classification, requiring less data and producing results that make chemical sense.
IAIFI Research Highlights
This work fuses condensed matter physics (knowledge of the periodic table, electronic topology, and metallicity) with machine learning methodology, producing models that are simultaneously predictive and physically interpretable.
The chemistry-informed inductive bias framework demonstrates a general principle: encoding domain structure into model architecture reduces data requirements, a transferable insight for AI in any data-scarce scientific domain.
By extending the topogivity approach to metallicity and providing a systematic framework for heuristic rules, this work offers new tools for rapidly screening and discovering materials with targeted electronic properties, including topological phases.
Future work could explore richer periodic-table embeddings and extension to structure-dependent properties; the paper is available at [arXiv:2505.02361](https://arxiv.org/abs/2505.02361) and builds directly on the topogivity work introduced by Ma et al.