
Machine learning electronic structure and atomistic properties from the external potential
Authors
Jigyasa Nigam, Tess Smidt, Geneviève Dusson
Abstract
Electronic structure calculations remain a major bottleneck in atomistic simulations and, not surprisingly, have attracted significant attention in machine learning (ML). Most existing approaches learn a direct map from molecular geometries, typically represented as graphs or encoded local environments, to molecular properties or use ML as a surrogate for electronic structure theory by targeting quantities such as Fock or density matrices expressed in an atomic orbital (AO) basis. Inspired by the Hohenberg-Kohn theorem, in this work, we propose an operator-centered framework in which the external (nuclear) potential, expressed in an AO basis, serves as the model input. From this operator, we construct hierarchical, body-ordered representations of atomic configurations that closely mirror the principles underlying several popular atom-centered descriptors. At the same time, the matrix-valued nature of the external potential provides a natural connection to equivariant message-passing neural networks. In particular, we show that successive products of the external potential provide a scalable route to equivariant message passing and enable an efficient description of long-range effects. We demonstrate that this approach can be used to model molecular properties, such as energies and dipole moments, from the external potential, or learn effective operator-to-operator maps, including mappings to the Fock matrix and the reduced density matrix from which multiple molecular observables can be simultaneously derived.
Concepts
The Big Picture
Imagine predicting how a molecule will behave without running expensive quantum chemistry calculations. Will it react? What color light does it absorb? How might it bind to a drug target? Most ML approaches treat molecules as graphs or point clouds and learn to map geometry to properties. But quantum mechanics has been hinting at a smarter starting point for decades.
That starting point is the external potential: the electrostatic pull that atomic nuclei exert on electrons. In the 1960s, Hohenberg and Kohn proved that this potential alone contains everything needed to determine a molecule’s lowest-energy state. Every energy level, every reaction rate, every optical property is encoded in how the nuclei push and pull on the electron cloud. Machine learning researchers at MIT and the Université Marie et Louis Pasteur have now built a framework that takes this theorem seriously as an engineering principle, not just a theoretical curiosity.
Jigyasa Nigam, Tess Smidt, and Geneviève Dusson propose treating the external potential, encoded as a matrix capturing how every nucleus influences the electrons, as the fundamental input to their ML models (arXiv:2602.15345). The result is a unified framework that predicts molecular properties, learns key quantum mechanical operators, and respects the symmetries that physics demands.
Key Insight: By grounding molecular ML in the Hohenberg-Kohn theorem and using the external potential matrix as input, this framework inherits both the theoretical rigor of density functional theory and the flexibility to learn any quantum mechanical observable.
How It Works
The core move is a change of representation. Instead of encoding a molecule as a graph of atoms and bonds, the team encodes it as a matrix V, the external potential matrix expressed in an atomic orbital (AO) basis. These are standard mathematical functions centered on each atom that describe electron distributions. Each matrix element captures how strongly two basis functions “feel” the nuclear attraction from every atom, giving a compact, information-rich fingerprint of the entire electrostatic environment.
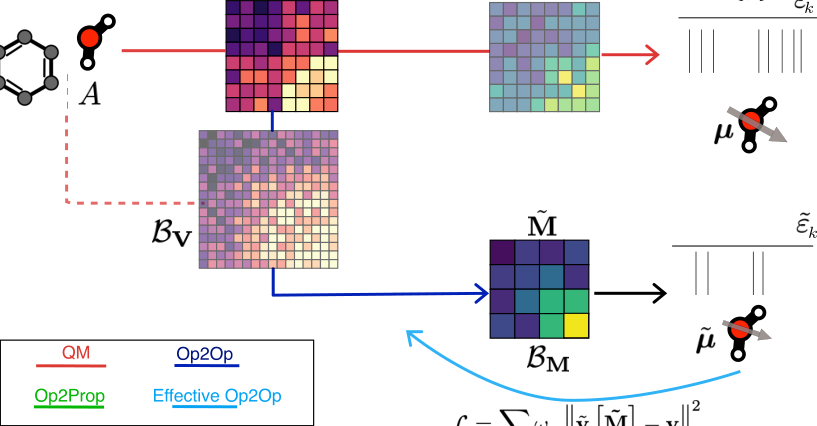
From this matrix, the team builds increasingly complex features through a single elegant operation: matrix multiplication. Each successive power of V captures higher-order correlations (two-body, three-body, four-body atomic interactions), mirroring the body-ordered atom-centered descriptors that are standard currency in molecular ML. SOAP (Smooth Overlap of Atomic Positions) and ACE (Atomic Cluster Expansion) are the two most widely used frameworks built on exactly these descriptors. The matrix structure of V generates all of this naturally, without separate engineering of higher-order terms.
This matrix multiplication route also connects directly to equivariant message-passing neural networks (MPNNs), the current state of the art for molecular ML. “Equivariant” here means the network’s outputs transform correctly under rotation or reflection: rotate the input molecule, and the predicted properties rotate with it. The team shows that multiplying external potential matrices achieves the same information propagation as MPNN message-passing, with each matrix product carrying atomic interactions one bond-length further. Rotational symmetry comes along for free through the mathematical structure of the AO basis.
Two model classes emerge from this:
- Op2Prop (Operator-to-Property): predicts scalar or vector quantities like energies and dipole moments directly from V
- Op2Op (Operator-to-Operator): learns to map V to other quantum mechanical operators, such as the Fock matrix or the reduced density matrix
The Op2Op variant is particularly powerful. The Fock matrix encodes how each electron effectively experiences both the nuclei and the averaged influence of all other electrons; it sits at the center of most quantum chemistry calculations. The reduced density matrix encodes how electrons are distributed and correlated across the molecule. Once you have a predicted Fock matrix, you can diagonalize it to get orbital energies and derive multiple molecular observables simultaneously, all from a single model.
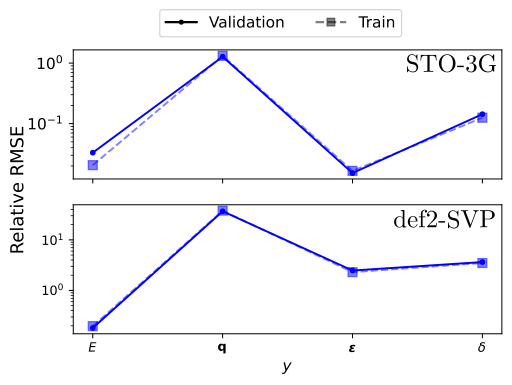
The team also introduces an effective Op2Op mode, where the model is supervised on physical observables like eigenvalues rather than matrix elements directly. This opens the door to learning from high-accuracy reference data without needing matched-basis calculations.
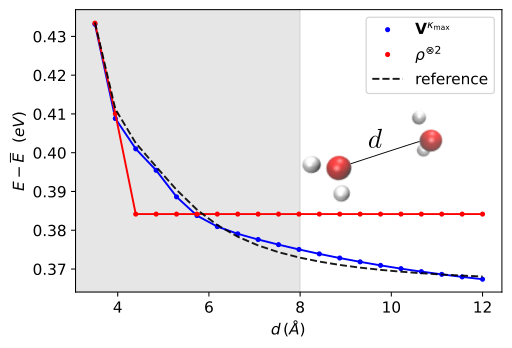
One persistent challenge for molecular ML is long-range interactions, electrostatic effects stretching across a molecule or between distant functional groups. Graph-based approaches handle this poorly because they rely on local neighborhoods. The external potential matrix sidesteps this by design: every matrix element involves contributions from all nuclei, so long-range Coulomb interactions are baked in from the start. Successive matrix products propagate this non-local information through the feature hierarchy without any special architectural tricks.
Why It Matters
The broader significance here is unification. Electronic structure ML currently comprises a menagerie of specialized models: some predict energies, some predict electron densities, some learn Hamiltonians. This framework offers a single input representation, the external potential matrix, from which all of these targets can be learned within a consistent theoretical language. Models trained for one task can share representations with models trained for another, and the Hohenberg-Kohn theorem provides a principled foundation for what the models can and cannot learn.
For the AI side, the connection to equivariant message-passing networks could flow in both directions. The external potential framework gives a physically motivated derivation of why message-passing works for molecules. It also suggests new architectural choices grounded in operator algebra rather than graph heuristics. As models scale to larger systems and more complex properties, that theoretical anchor could prove decisive.
Open questions remain. The AO basis used to discretize the input is a tunable hyperparameter, and choosing it wisely (balancing resolution against computational cost) is an art the paper begins to formalize but doesn’t fully resolve. Scaling to very large molecules, where the full external potential matrix becomes expensive to compute and store, will require further innovation. The effective Op2Op approach also opens a door toward models that learn from experimental data or high-level theory without requiring matched-basis reference calculations, a direction worth watching.
Bottom Line: By grounding molecular machine learning in the Hohenberg-Kohn theorem and treating the external potential matrix as the fundamental input, this work delivers a unified, symmetry-aware framework for predicting both molecular properties and quantum mechanical operators, with long-range physics included for free.
IAIFI Research Highlights
Interdisciplinary Research Achievement: The work connects the theoretical foundations of density functional theory, specifically the Hohenberg-Kohn theorem, with modern equivariant machine learning architectures. Quantum mechanical principles directly motivate the model design choices rather than being imposed after the fact.
Impact on Artificial Intelligence: The paper establishes a rigorous connection between external potential matrix products and equivariant message-passing neural networks. This gives a physically grounded explanation for why graph-based molecular ML works and points toward new operator-algebra-based architectures.
Impact on Fundamental Interactions: By learning operator-to-operator maps (including Fock and density matrices), the framework enables simultaneous prediction of multiple quantum mechanical observables from a single model, potentially accelerating electronic structure calculations across chemistry and materials science.
Outlook and References: Scaling to large molecules and refining basis-set hyperparameter choices are the most pressing next steps. The paper appeared on arXiv:2602.15345 from MIT’s Research Laboratory of Electronics and the Université Marie et Louis Pasteur.