
Predicting band gap from chemical composition: A simple learned model for a material property with atypical statistics
Authors
Andrew Ma, Owen Dugan, Marin Soljačić
Abstract
In solid-state materials science, substantial efforts have been devoted to the calculation and modeling of the electronic band gap. While a wide range of ab initio methods and machine learning algorithms have been created that can predict this quantity, the development of new computational approaches for studying the band gap remains an active area of research. Here we introduce a simple machine learning model for predicting the band gap using only the chemical composition of the crystalline material. To motivate the form of the model, we first analyze the empirical distribution of the band gap, which sheds new light on its atypical statistics. Specifically, our analysis enables us to frame band gap prediction as a task of modeling a mixed random variable, and we design our model accordingly. Our model formulation incorporates thematic ideas from chemical heuristic models for other material properties in a manner that is suited towards the band gap modeling task. The model has exactly one parameter corresponding to each element, which is fit using data. To predict the band gap for a given material, the model computes a weighted average of the parameters associated with its constituent elements and then takes the maximum of this quantity and zero. The model provides heuristic chemical interpretability by intuitively capturing the associations between the band gap and individual chemical elements.
Concepts
The Big Picture
Imagine predicting how spicy a dish will be. You could use a lab spectrometer to measure capsaicin concentrations (precise, but slow and expensive), or you could scan the ingredient list: chilies and black pepper push spiciness up, cream and sugar push it down. That rule of thumb won’t win a culinary science prize, but it works surprisingly well for a quick assessment. Now apply the same logic to one of the most important properties in materials science.
The electronic band gap determines whether a material conducts electricity like copper, blocks it like glass, or falls somewhere in between as a semiconductor like silicon. Getting it right matters enormously for designing solar cells, transistors, and LEDs. For decades, scientists have calculated band gaps using expensive quantum mechanical simulations, or trained complex machine learning models that require knowing a material’s full crystal structure (the precise arrangement of every atom). But what if a surprisingly good estimate required only the chemical formula?
Researchers at MIT have done exactly that, and in the process discovered something unexpected about the band gap’s statistics.
Key Insight: The band gap is not a simple continuous quantity. It behaves as a mixed random variable, with a finite fraction of materials landing at exactly zero. Recognizing this statistical quirk unlocks a cleaner, more interpretable model.
How It Works
The first breakthrough isn’t the model. It’s the observation that motivated it. Most machine learning approaches treat band gap prediction as standard regression: predict a number. Regression assumes a smooth, continuous target (like height or temperature, which can take any value on a sliding scale), but the band gap doesn’t cooperate.
The MIT team plotted the empirical cumulative distribution function (eCDF) of band gaps across thousands of materials, a graph showing what fraction of materials fall at or below each possible value. What they found was striking.
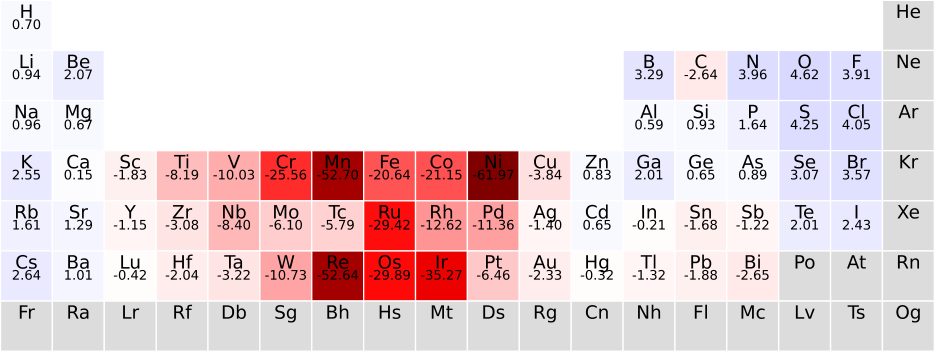
The distribution has a sharp jump at exactly 0 eV (electron volts, the standard energy unit for atomic-scale physics). A large fraction of crystalline materials are metals, meaning their band gap is exactly zero. The rest (semiconductors and insulators) have band gaps spread continuously from small positive values up to around 10 eV or more.
Formally, the band gap is a mixed random variable: it partly snaps to a fixed value (exactly zero, for metals) and partly varies continuously (any positive value, for semiconductors and insulators). Train a standard regression model on this data and you implicitly allow predictions of slightly negative or near-zero values that have no physical meaning.
The model the team designed respects this structure:
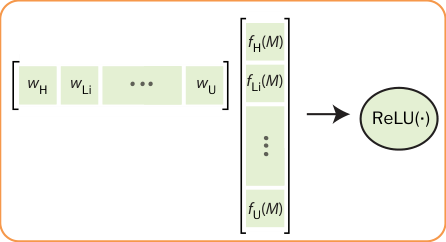
- One parameter per element. Every element in the periodic table gets a single learned number, its “band gap tendency,” fit from data.
- Weighted average. For a given material, compute a weighted average of those parameters using the subscripts in the chemical formula as weights (in BaTiO₃, barium and titanium each contribute weight 1, oxygen contributes weight 3).
- ReLU activation. Take the maximum of that weighted average and zero.
That final step, the ReLU (Rectified Linear Unit) borrowed from neural network architecture, encodes the mixed-variable nature of the band gap directly. A negative weighted average predicts zero: a metal. A positive weighted average predicts that value: a semiconductor or insulator. The hard floor at zero isn’t a trick; it flows directly from the statistical analysis.
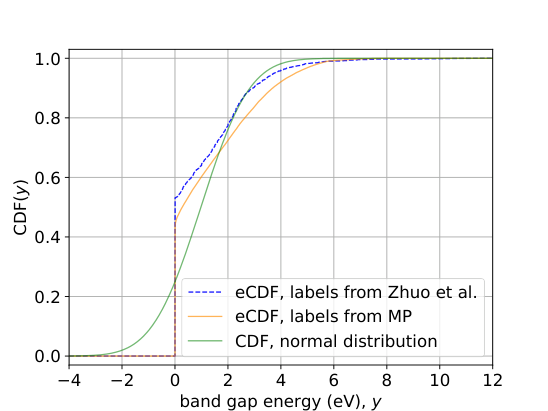
The model’s interpretability is genuine, not retrofitted. Elements with high parameter values (fluorine, oxygen) tend to appear in ionic compounds with large band gaps. Elements with low or negative values (iron, platinum) tend to appear in metallic compounds. The learned parameters match known chemical intuition about which elements drive insulating versus metallic behavior.
Why It Matters
The immediate practical advantage is accessibility. Quantum simulation methods such as density functional theory (DFT) and graph neural networks both require knowing a material’s crystal structure (the coordinates of every atom in the repeating structural unit), and that information isn’t always available when screening hypothetical or poorly characterized materials. A model that works from chemical formulas alone can operate earlier in the discovery pipeline, flagging promising candidates before expensive structural calculations begin.
There’s also a deeper point about modeling philosophy. The materials science community has accumulated decades of chemical intuition: which elements promote ionic versus metallic bonding, how electronegativity (how strongly each element attracts electrons) relates to band gaps, which structural families tend to produce semiconductors. This model sits within that tradition. Simple enough to be fully understood, expressive enough to be useful, and structured so its parameters translate directly into chemical insight.
Black-box neural networks dominate the field, so this represents a deliberate trade: some predictive ceiling for total interpretability. The authors argue that trade-off is worth it for compositional screening, and the approach opens the door to similar element-parameter models for other material properties where composition dominates structure.
Bottom Line: By recognizing that the band gap is statistically mixed (not purely continuous), this team built a one-parameter-per-element model that is transparent, fast, and formula-accessible, offering a principled alternative to black-box ML for materials screening.
IAIFI Research Highlights
This work applies statistical analysis and machine learning design principles to a core problem in condensed matter physics, producing a model whose architecture is directly motivated by the quantum mechanical reality of metallic versus insulating materials.
Careful statistical characterization of a target variable (recognizing it as a mixed random variable rather than a continuous one) can improve model design more than adding complexity. That's a lesson that extends well beyond materials science.
By assigning a single learned parameter to each chemical element, the model recovers and quantifies known chemical heuristics about which elements promote or suppress the electronic band gap, giving a data-driven handle on elemental chemistry.
Future work could extend this element-parameter framework to other material properties with atypical statistics, and explore how compositional models complement structural models in high-throughput discovery pipelines. Full paper: [arXiv:2501.02932](https://arxiv.org/abs/2501.02932)